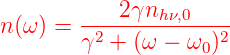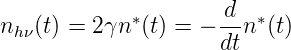

| Versuch zur Vorlesung: | |
| Linienspektren: Quecksilber, Helium, Kalium, Cadmium, Krypton, Zink (Versuchskarte AT-46) | |
Wenn ein Atom in einem angeregten Zustand ist, emittiert es seine Energie als Photon zu einem zufälligen Zeitpunkt. Wenn n∗ die Anzahl der angeregten Atome ist, dann ist die Rate nhν der ausgesandten Photonen in den Raum (Ω = 4π)
|
| (7.1) |
proportional zur Anzahl der angeregten Atome n∗. Die Lösung von Gleichung (7.1) ist eine Exponentialfunktion. Mit der Anfangsbedingung n∗(0) = n 0∗ = n hν(0)∕(2γ) = nhν,0∕(2γ) erhalten wir für die Anzahl angeregter Atome
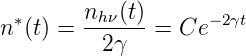 | (7.2) |
und daraus die Anzahl Photonen pro Zeit
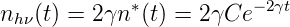 | (7.3) |
Die Gesamtzahl aller emittierten Photonen ist
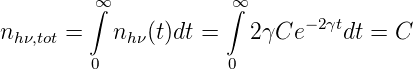 | (7.4) |
Andererseits folgt aus (7.2), dass die Anzahl der angeregten Atome zur Zeit t = 0 gerade n∗(0) = C = n 0∗ ist. Wir erhalten also Normierungskonstante
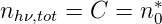 | (7.5) |
Damit ist die Gesamtzahl aller ausgesandter Photonen gleich der Zahl der angeregten Atome n∗(0) = n 0∗ zur Zeit t = 0.
Die Lebensdauer des Zustandes ist τ = 1∕(2γ). Sie hängt mit dem Einstein-Koeffizienten A der spontanen Emission (aus Gleichung (3.31)) zusammen.
Die Anzahl Photonen ist proportional zur Energie. Die Rate mit der die Energie abnimmt ist das Quadrat der Rate, mit der die Amplitude abnimmt. Die Amplitudenabnahme ist
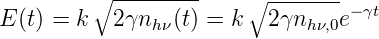 | (7.6) |
k ist eine Proportionalitätskonstante, die wir hier nicht bestimmen. Aus diesem Verlauf der Amplitude Gleichung (7.6) kann die Linienform über eine einseitige Fouriertransformation F oder eine Laplacetransformation L berechnet werden. Man erkennt, dass wir eine Linie um die Mittenfrequenz ω0 haben. Deshalb wird anschliessend s → i(ω −ω0) ersetzt (Die Überlegung ergibt somit die Frequenzabweichung, nicht die Frequenz). Vom Resultat wird das Betragsquadrat gebildet. Die einseitige Fouriertransformation wird benötigt, da die zweiseitige Fouriertransformation nicht kausal ist.
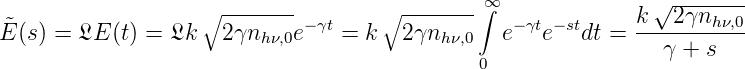 | (7.7) |
Wir ersetzen nun in Gleichung (7.7) s mit i(ω −ω0)
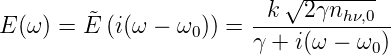 | (7.8) |
Wir suchen aber die Linienform der Intensität (Energie pro Fläche und Zeit). Deshalb müssen wir das Betragsquadrat berechnen.
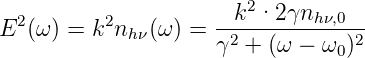 | (7.9) |
Wir erhalten damit die Form der
Lorentzlinie
|
Unsere Normierung garantiert, dass wir wieder die Gesamtzahl der angeregten Atome erhalten, sofern wir über die Frequenz ν = ω∕(2π) integrieren.
__________________________________________________________________________
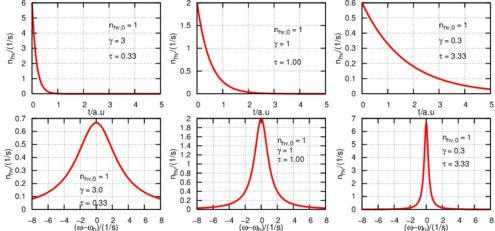
Zusammenhang zwischen der Abfallzeit und der Breite der Lorentzlinie. Oben sind die Abfallzeiten gezeigt, darunter die dazu gehörigen Linienbreiten.
_____________________________________________________________________
Abbildung 7.1.1 zeigt einige Abfallkurven für die Energie (oder Intensität) und die dazugehörigen Linienbreiten. Wenn die Lebensdauer zunimmt, nimmt die Linienbreite ab.
Typische Werte für die Lebensdauer τ sind 1 ns bis 10 ns. Entsprechend ist γ = 5·107 s−1 bis 5·108 s−1. Diese Lebensdauer wird auch die natürliche Lebensdauer genannt.
Der Einstein-Koeffizient A der spontanen Emission (aus Gleichung (3.31)) gibt die Anzahl der Emissionen pro Volumen und Zeit an, wenn er mit der Anzahl der Atome im angeregten Zustand n∗ multipliziert wird.
| Versuch zur Vorlesung: | |
| Quecksilber: Druckverbreiterung von Spektrallinien (Versuchskarte AT-47) | |
Die natürliche Linienbreite ist durch die Lebensdauer des angeregten Zustandes τ gegeben. Diese Lebensdauer kann aus der Grösse der Übergangsmatrixelemente zwischen den Elektronenzuständen berechnet werden. Aus dem Abschnitt über den Lorentz-Oszillator wissen wir, dass die Lebensdauer des Zustandes τ = 1∕(2γ) ist. Mit der Gleichung (7.10) bekommen wir
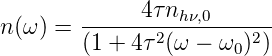 | (7.11) |
Dies ist die natürliche Linienbreite.
Die Atome sind in der Regel nicht in Ruhe. Sie bewegen sich auf den Beobachter zu oder weg. Da die Geschwindigkeiten klein gegen die Lichtgeschwindigkeit sind, kann mit der linearisierten Dopplerverschiebung gerechnet werden. Ohne Einschränkung der Allgemeinheit legen wir die Beobachtungsrichtung in die x-Richtung
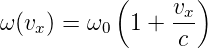 | (7.12) |
Für die beobachtete Dopplerverschiebung ist nur die Komponente vx in Richtung der Wellenausbreitung wichtig. Der Geschwindigkeitsbetrag der Atome in Richtung des Beobachters ist (Siehe Reif, Statistical and Thermal Physics [Rei65, pp. 265])
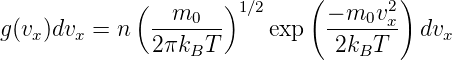 | (7.13) |
wichtig. Aus der Dopplerverschiebung erhalten wir
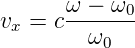 | (7.14) |
Eingesetzt in Gleichung (7.13) ergibt sich
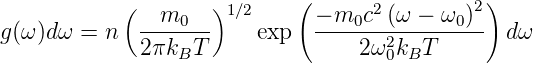 | (7.15) |
Diese Verteilungsfunktion muss auch in der Intensität zu finden sein, wobei wir eine unspezifizierte Konstante C verwenden.
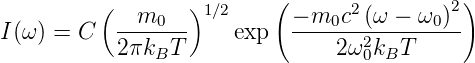 | (7.16) |
Die für die Linienform charakteristische Grösse ist die Halbwertsbreite Δω1∕2 = ω+1∕2 −ω−1∕2 definiert als I(ω±1∕2) = I(ω0)∕2.
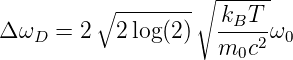 | (7.17) |
Zum Beispiel sind die Natrium D-Linien bei etwa 589 nm. Dies entspricht einer Frequenz von νNaD = 5.08985·1014 s−1. Ein Natriumatom hat eine Masse von m0 = 22.98976928 u = 3.81754·10−26 kg. Bei einer Temperatur von 1273 K ist die Dopplerverbreiterung
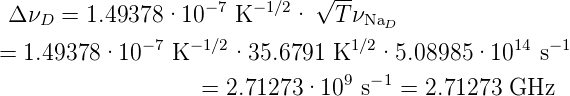 |
Gleichung (7.16) kann für Na auch so geschrieben werden:
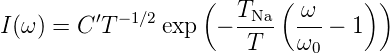 |
mit TNa = 1.24254·1014 K. Abbildung 7.1.2.2 zeigt einen Vergleich der Dopplerverbreiterung bei 1273 K und 273 K mit der natürlichen Linienbreite bei einer Lebensdauer von τ = 16.3 ns.
__________________________________________________________________________
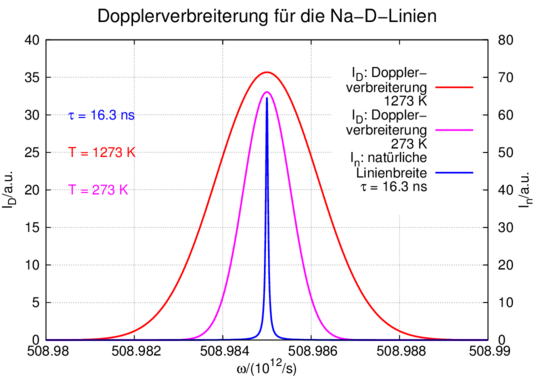
Vergleich der Dopplerverbreiterung mit der natürlichen Linienbreite bei den Natrium-D-Linien (entspricht dem Falle T = 0 K).
_____________________________________________________________________
Der Einstein-Koeffizient A der spontanen Emission (aus Gleichung (3.31)) gibt die Anzahl der Emissionen pro Volumen und Zeit, wenn er mit der Anzahl der Atome im angeregten Zustand n∗ multipliziert wird.
__________________________________________________________________________
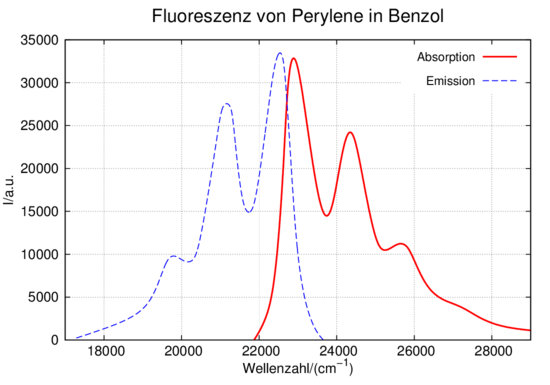
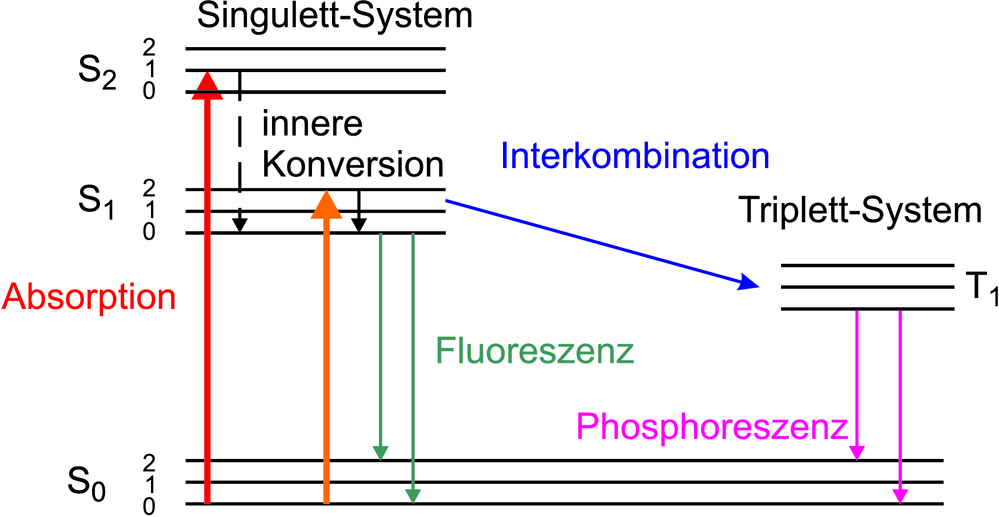
Fluoreszenz von Perylene in Benzol (adaptiert aus [Lak06] und rechts: Jablonski-Diagramm
_____________________________________________________________________
Abbildung 7.1.3 zeigt ein Fluoreszenzspektrum von Perylene in Benzol und auf der rechten Seite das dazugehörige Jablonski-Diagramm. Die Fluoreszenz wurde von William Herschel im Jahre 1845 entdeckt. Sie spielt sich im Singulett-System ab. Die einzelnen Niveaus in ausgedehnten Molekülen sind durch Vibrationen und Rotationen noch weiter aufgespalten. Die Anregung erfolgt dann vom Grundzustand des Singulett-Systems in einen der vibratorisch angeregten Zustände eines höheren Quantenniveaus. Durch die Druckverbreiterung bilden diese Vibrationszustände ein Quasikontinuum. Nach dem Franck-Condon-Prinzip ändert ein Elektron bei der Anregung (dauert etwa 10−15 s) in ein höheres Niveau seinen Ort kaum. Da die Minima der höheren energetischen Zustände weiter vom Kern weg sind, endet das Elektron üblicherweise in einem vibratorisch angeregtes Niveau. Es relaxiert dann im Mittel innert weniger als einer Picosekunde ind den vibratorischen Grundzustand des angeregten Niveaus. Im Mittel bleibt dann das Elektron während der Fluoreszenzlebensdauer (10−9 s bis 10−8 s) im höheren Niveau um dann unter Aussendung eines Fluoreszenzphotons in einen der vibratorisch angeregten Unterzustände des Grundzustandes zu gelangen. Durch diese Prozesse entsteht ein Stokes-Shift. Dieser zeigt sich in Abbildung 7.1.3 darin, dass das Absorptionsspektrum bei höheren Energien ist als das Emissionsspektrum.
Die Abstände der Vibrationsniveaus sind etwa gleich im Grundzustand wie in den angeregten Zuständen. Das führt dazu, dass das Emissionsspektrum und das Absorptionsspektrum oft spiegelbildlich liegen (Kasha’s Regel). Diese Regel gilt nicht, wenn ein Molekül bei der Absorption eines Photons ionisiert wird oder wenn es seine Konformation ändert.
Wenn der Übergang in den S2-Zustand angeregt wird, relaxiert er ziemlich schnell in den S1-Zustand. Sowohl da wie auch bei der vibratorischen Abregung werden Energie- und Impulserhaltung durch den Rest des Moleküls oder oft durch die umgebende Flüssigkeit garantiert.
Nach der Auswahlregel aus Gleichung (6.16) koppeln das Singulett- und das Triplettsystem nicht. In einem komplizierten Molekül ist es aber nicht unwahrscheinlich, dass der durch die Impuls- und Drehimpulserhaltung verbotene Übergang von S2 nach T1 in Abbildung Gleichung (7.1.3) mithilfe eines dritten Partners möglich ist. Wenn der Übergang durch Interkombination (auch Intersystem Crossing)in den Triplettzustand gelangt ist, muss er in diesem metastabilen Zustand wesentlich länger als die Fluoreszenzlebensdauer von etwa τ ≈ 10−8 s bleiben. Die mittlere Lebensdauer eines Triplettzustandes T1 beträgt von Millisekunden bis Stunden. Selbstleuchtende Uhrzeiger sind heute phosphoreszierend. Früher wurde die Fluoreszenz durch radioaktive Stoffe erzeugt. Beim Übergang von T1 → S1 tritt wieder Interkombination auf.
Beim Ramaneffekt (erstmals beobachtet 1928 von Chandrasekhara Raman) gewinnt oder verliert das Licht Energie bei der Streuung an einem Atom oder Molekül. Die übliche Streuung von Licht (Rayleigh-Streuung) kann quantenmechanisch so verstanden werden, dass ein Photon aus dem Grundzustand S0 (siehe Abbildung 7.1.5) in ein virtuelles Niveau angeregt wird und dann spontan (Einstein-Koeffizient A) wieder in eine beliebige Richtung emittiert wird. Aus der Unschärferelation für Energie und Zeit ersieht man, dass
 | (7.18) |
sein muss. Das heisst, für ganz kurze Zeiten (der Effekt ist nicht messbar, aber die Auswirkungen beobachtbar) kann ein virtuelles Niveau existieren. Im Falle von sichtbarem Licht heisst das, dass Δt ≈ 1 fs sein muss.
Wir hatten bei der Fluoreszenz gesehen, dass das Grundniveau S0 vibratorisch aufgespalten ist. Wir könnten mit Infrarotlicht den vibratorischen Übergang mit hνv direkt ansprechen und spektroskopieren. Bis vor kurzem waren aber keine durchstimmbaren Laser in diesem Wellenlängenbereich vorhanden. Zudem ist die Optik unhandlicher und teurer.
__________________________________________________________________________
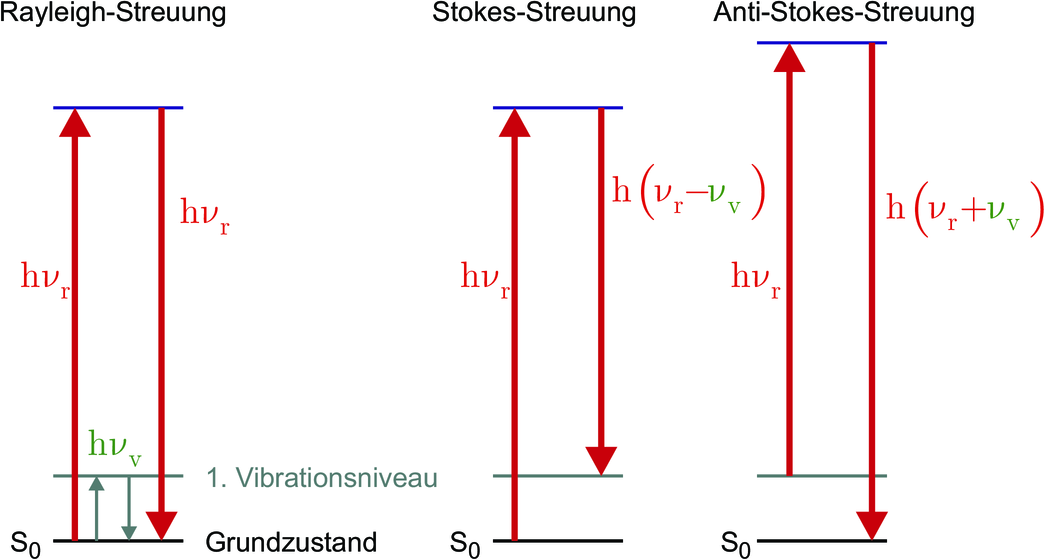
Illustration der Ramanstreuung im Vergleich zur Rayleighstreuung.
_____________________________________________________________________
In Abbildung 7.1.5 in der Mitte ist der Stokes-Shift angegeben. Das Elektron im virtuellen Niveau kann auch in das erste vibratorisch angeregte niveau relaxieren. Seine Frequenz ist dann
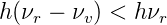 | (7.19) |
kleiner als das eingestrahlte licht. Wir erwarten also, dass wir auf der längerwelligen Seite der Rayleigh-Linie eine schwache Linie im Abstand der Vibrationsfrequenz des ersten Vibrationsniveaus finden. Diese Linie wird beobachtet, sie ist aber um Grössenordnungen schwächer, da der Streuquerschnitt für Ramanstreuung nur etwa σRaman = 10−34 m2 beträgt.
Neben der Stokes-Linie gibt es noch die Anti-Stokes-Linie. Abbildung 7.1.5 rechts zeigt die Niveaus beim Anti-Stokes-Shift. Hier wird ein Elektron im ersten vibratorischen Niveau durch das einfallende Photon in ein virtuelles Niveau angeregt. Dieses relaxiert nachher in das Grundniveau. Wir haben also
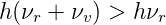 | (7.20) |
Hier hat das Raman-gestreute Photon eine höhere Frequenz und eine kleinere Wellenlänge als das einfallende Photon. Da das erste vibratorisch angeregte Niveau nach der Boltzmannstatistik weniger stark besetz ist als der Grundzustand, ist die Anti-Stokes-Linie schwächer als die Stokes-Linie.
__________________________________________________________________________
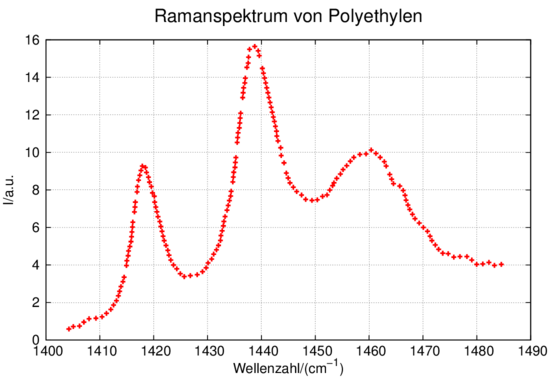
Ramanspektrum von Poyletylen im Bereich der Methylen-Biegeschwingung (adaptiert aus [BK70])
_____________________________________________________________________
Abbildung 7.1.5 zeigt beispielhaft ein Ramanspektrum von kristallinem Polyethylen (siehe auch [BK70]). Die Auswahlregeln für Dipolübergänge sagen, dass die Übergänge, die in der Infrarotspektroskopie beobachtet werden, also den üblichen Auswahlregeln für optische Spektroskopie genügen, nicht in Ramanspektren beobachtet werden, und umgekehrt.
 Lizenzinformationen
Lizenzinformationen