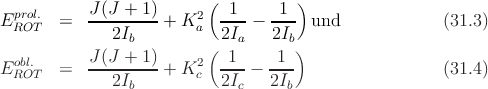
| Abbildung 31.1: | Termschema zu 3 |
MODR ist ein modernes spektroskopisches Verfahren, mit dem man rovibronische energetische Strukturen freier Moleküle, also die Quantenstrukturen der Elektronen-, Schwingungs- und Rotationsfreiheitsgrade bis hin zur natürlichen Linienbreite auflösen kann. Es werden die hochfrequenten Quanten des Fluoreszenzlichts angeregter Elektronenzustände registriert, was in einer ungewöhnlich hohen spektralen Empfindlichkeit resultiert. Der Versuch eröffnet die Möglichkeit, die methodischen Tricks kennenzulernen, mit denen man mit zwei so unterschiedlichen kohärenten Feldern wie Mikrowellen und Laserlicht gleichzeitig auf ein Quantenobjekt einwirken kann, andererseits aber auch die aus der Quantenmechanik entlehnten Hilfsmittel, die die Meßergebnisse molekülphysikalisch umzusetzen gestatten.
Allgemein:
Speziell:
Meßmethodik:
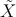 413leftarrow
413leftarrow 414 (10463.94MHz) auf und optimieren Sie
Signal-Rausch-Verhältnis und Linien-breite. Als Anhaltspunkt dienen Ihnen
die eingestellten Parameter vom ersten Versuchstag (Messprotokoll!).
414 (10463.94MHz) auf und optimieren Sie
Signal-Rausch-Verhältnis und Linien-breite. Als Anhaltspunkt dienen Ihnen
die eingestellten Parameter vom ersten Versuchstag (Messprotokoll!).
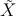 716 (16753.35cm-1), 41Ã7
07 ← X*
(8170.6MHz) bei den Auswahlregeln
716 (16753.35cm-1), 41Ã7
07 ← X*
(8170.6MHz) bei den Auswahlregeln
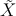 413 ←
413 ←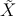 414 mit
der Laseranregung 41Ã3
03 ←
414 mit
der Laseranregung 41Ã3
03 ← 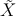 414 mit 16751.56cm-1 (Auswahlregeln:
ΔMJ(MW) = ±1, ΔMJ(Laser) = 0 ). Nehmen Sie wenigstens fünf Spektren
zwischen 0T und 1.3T auf.
414 mit 16751.56cm-1 (Auswahlregeln:
ΔMJ(MW) = ±1, ΔMJ(Laser) = 0 ). Nehmen Sie wenigstens fünf Spektren
zwischen 0T und 1.3T auf.
ACHTUNG: Das im Praktikum verwendete Lasersystem (Pump- und Farbstofflaser) gehört der Laserklasse IV an. Das bedeutet, dass bereits indirekte Strahlung (Reflexionen und Streuung) des Lasers die Augen irreparabel schädigen können. Tragen Sie deshalb unbedingt die bereitliegenden Schutzbrillen, wenn der Laser in Betrieb ist.
Frequenzänderungen des Laserlichts dürfen grundsätzlich nur vom Assistenten vorgenommen werden.
Vor Einschalten des Elektromagneten ist sicherzustellen, das die Wasserkühlung in Betrieb ist.
In diesem Versuch wird die gleichzeitige resonante Einwirkung zweier oszillierender E-Felder auf ein molekulares System untersucht (Doppel-Resonanz, DR) mit dem Ziel Informationen über dessen Energiezustände zu erlangen.
Die Schwingunsfrequenzen der beiden Felder sind stark verschieden, sie liegen im sichtbaren (Optischen) und im Mikrowellenbereich (MW). Der optische Anteil wird hier über einen Farbstoff-Ringlaser, der niederfrequente über einen Mikrowellengenerator im sog. X-Band (8.2-12.4 GHz) mit nachgeschaltetem Verstärker bereitgestellt. Über Farbstofflaser und auch den prinzipiellen experimentellen MODR-Aufbau kann man in Demtröder [1] nachlesen, nähere Auskunft über den Umgang mit Mikrowellen in der Spektroskopie geben Townes und Schawlow [2].
Die MODR ist ein hochauflösendes Verfahren, mit dem man die Dopplerverbreiterung der Sprektrallinien unterdrücken kann, die in optischen Übergängen dominiert. Konkurrierende Verbreiterungsmechanismen sind Wandstöße, Stöße zwischen Molekülen (Druckverbreiterung), Leistungssättigung und natürliche Lebensdauer. Diese Stichpunkte kann man in [2] oder vorzugsweise in Gordy und Cook [3] vertiefen.
Um sich mit dem Assistenten verständigen zu können, ist es unumgänglich, die Nomenklatur der Energieniveaus des untersuchten Moleküls zu kennen (es ist dies z.Zt. Thioformaldehyd, H2C = S, ein leicht asymmetrischer Rotator).
Die etwas schwierige Rotationsquantelung ist in einem eleganten, für Physiker besonders geeigneten Zugang in einem Artikel von Clasine van Winter [4] bearbeitet worden. Jedoch ist es für den Anfänger sehr zeitaufwendig, zu einem tiefgehenden Verständnis zu gelangen. Wir geben deshalb unten einige Kapitel aus [3] an, deren Lektüre zumindest das Verständnis dafür erleichtert, worauf die Nomenklatur einzelner Rotationsniveaus, JKaKc, basiert und wie man damit umgeht. Ausgangspunkt ist die Quantisierung des symmetrischen Kreisels, die einfach und in geschlossener Form möglich ist.
Man muss eigentlich lediglich wissen, dass es langgezogene (prolate) und abgeplattete (oblate) symmetrische Kreisel gibt. Verabredungsgemäß bezeichnet man die drei zueinander senkrechten Hauptachsen in der Abfolge zunehmenden Trägheitsmoments eines beliebigen Rotators mit a, b, c. Es gilt dann Ia < Ib = Ic für die Trägheitsmomente des prolaten, Ia = Ib < Ic für die des oblaten symmetrischen Grenzfalls (siehe Abb.1). Der Hamiltonoperator für den allgemeinen starren Kreisel lautet
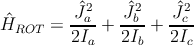 | (31.1) |
wobei Ĵi,i = a,b,c die Komponenten des Gesamtdrehimpulses bedeuten. Die genannten Schwierigkeiten entstehen dadurch, dass die Komponenten untereinander nicht vertauschen. In den Grenzfällen erhält man aber einfach (Ĵ2 = Ĵ a2 + Ĵ b2 + Ĵ c2)
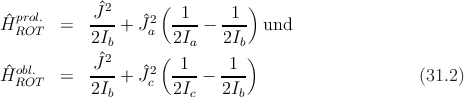
Da Ĵ2 mit sich und seinen Komponenten vertauscht, sind die beiden
Energieoperatoren diagonal in den Quantenzahlen J und Ka (bzw. J und Kc), wobei
Ka, Kc jeweils die Quantenzahlen der Projektion von 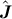 auf die Symmetrieachse
benennt, die ja eine Konstante der Bewegung sein muss. Für die Energie folgt
demnach
auf die Symmetrieachse
benennt, die ja eine Konstante der Bewegung sein muss. Für die Energie folgt
demnach
wobei Ka, Kc die 2J + 1 ganzzahligen Werte zwischen -J und +J annehmen.
Lässt man nun in Gedanken das mittlere Trägheitsmoment (Ib) von Ia nach Ic variieren, so erfasst man alle asymmetrischen Kreisel. Die so entstehenden ”Energiekurven” haben eine Struktur wie in Abbildung 31.2.
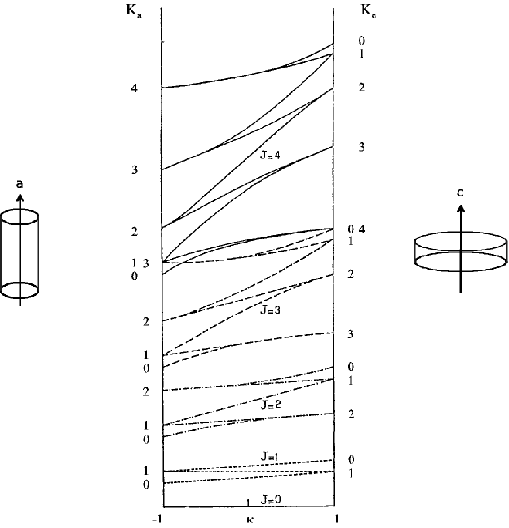
| Abbildung 31.2: | Energien des asymmetrischen Kreisels in Abhängigkeit des Asymmetrieparameters κ. Links stehen die K-Quantenzahlen des prolaten, rechts des oblaten symmetrischen Grenzfalls (aus [4]). Man überzeuge sich, dass die Energien an der linken Ordinate gemäß Gl. (31.3), an der rechten gemäß Gl. (31.4) strukturiert sind. |
Als variierenden Parameter haben wir nicht Ib, sondern den Asymmetrieparameter nach Ray,
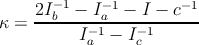 | (31.5) |
verwendet, der alle Werte zwischen -1 und +1 annehmen kann.
Den letzten Schritt zum Verständnis der Nomenklatur bzw. der Abzählweise der Rotatorniveaus kann man zurücklegen, wenn man die Tatsache entgegennimmt, dass sich die 2J + 1 Energiekurven in Abb. 31.2 für ein gegebenes J niemals schneiden. Es ist dann nämlich ein Energieniveau bei beliebigem κ durch Angabe von J und von Ka und Kc eindeutig festgelegt! Jeder weiß damit Bescheid, welches Niveau gemeint ist, das wir unter JKaKc ansprechen (z.B. sind 101, 111 und 110 die einzigen 2J + 1 = 3 Niveaus, die im Falle J = 1 möglich sind).
Die Eigenfunktionen des symmetrischen Kreisels hängen von den drei Eulerwinkeln ab und spannen einen vollständigen Hilbertraum auf. Technisch bewältigt man die Aufgabe der numerischen Berechnung der Energieeigenwerte eines asymmetrischen Kreisels deshalb so, dass man die Matrixelemente des Hamiltonoperators (Gl. (1)) in Abhängigkeit von Ia, Ib und Ic in der Basis des symmetrischen Kreisels aufstellt und die so gebildete Matrix dann diagonalisiert. Da der Hamiltonoperator Gl. (31.1) gegen die Operationen der sog. Vierergruppe (E,C2a,C 2b,C 2c) invariant ist, kann man Symmetrieüberlegungen vorteilhaft nutzen [4].
Der Vollständigkeit halber sei noch erwähnt, dass auch Ĵz, der Operator der
Projektion von 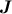 auf die laborfeste Achse Z, mit ĤROT in Gl. (31.1) kommutiert.
Daraus resultieren die M-Zustände mit M als der magnetischen Quantenzahl. Dies wird
aber nur wichtig im Falle äußerer Felder. Im feldfreien Raum sind die 2J + 1
verschiedenen M-Zustände energetisch entartet und fallen mit den oben besprochenen
Nieveaus JKaKc zusammen.
auf die laborfeste Achse Z, mit ĤROT in Gl. (31.1) kommutiert.
Daraus resultieren die M-Zustände mit M als der magnetischen Quantenzahl. Dies wird
aber nur wichtig im Falle äußerer Felder. Im feldfreien Raum sind die 2J + 1
verschiedenen M-Zustände energetisch entartet und fallen mit den oben besprochenen
Nieveaus JKaKc zusammen.
Für eine nähere Auseinandersetzung mit den theoretischen Grundlagen dieses FP-Versuchs empfehlen wir, folgende Unterkapitel in Gordy und Cook [3] anzuschauen (insgesamt etwa 30 Seiten):
Zum Schluss sei noch betont, dass im MODR-Verfahren die Rotationsniveaus eines angeregten Elektronenzustandes mit denen des Grundzustandes verknüpft werden (zusätzlich können die Moleküle auch noch schwingen, was für uns aber nur am Rande von Bedeutung ist). Da sich die Struktur mit der Elektronenanregung im allgemeinen stark ändert, ändern sich natürlich auch die Trägheitsmomente und damit die Rotationsenergien bezüglich derer des Grundzustandes.