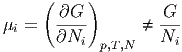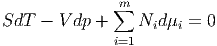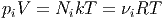

Bei idealen Gasen erfüllen die Teilchensorten das Volumen so, wie wenn sie es alleine ausfüllen würden. Bei idealen Gasen existieren keine langreichweitigen Kräfte. Die Stösse zwischen den unterschiedlichen Molekülsorten gleichen die Temperatur aus, so dass alle Molekülsorten die gleiche Temperatur haben. Für die i-te Teilchensorte können wir also schreiben:
|
| (2.1) |
Mit dem Daltonschen Gesetz (Gleichung (2.2)) lautet dann die Zustandsgleichung für Mischungen des idealen Gases
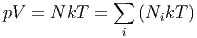 | (2.3) |
Die innere Energie dieses Gemisches ist
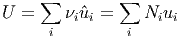 | (2.4) |
wobei
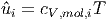 |
die innere Energie pro mol der Sorte i und
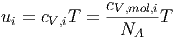 |
die mittlere innere Energie pro Molekül der Sorte i ist.
Die verschiedenen Molekülsorten des Gasgemisches nehmen den gleichen Raum ein. Die Moleküle wechselwirken, ausser bei Stössen, nicht miteinander. Deshalb sind die resultierenden Wärmekapazitäten die Summe aus den Wärmekapazitäten der einzelnen Teilsysteme, unabhängig davon ob V oder p konstant gehalten wird.
| CV | = ∑ iνicV ,mol,i | = | ∑
i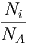 cV ,mol,i cV ,mol,i | (2.5) |
| Cp | = ∑ iνicp,mol,i | = | ∑
i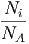 cp,mol,i cp,mol,i | (2.6) |
Dabei ist, bei genügend hohen Temperaturen
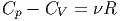 |
oder
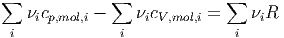 | (2.7) |
Da Gleichung (2.7) für alle Kombinationen von νi gelten muss, muss für jede einzelne Teilsorte des Gemisches
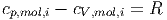 | (2.8) |
gelten.
Bei Gemischen haben wir für die obigen Betrachtungen angenommen, dass jede Molekülsorte das ganze Volumen V einnimmt, dass aber ihr Partialdruck pi ein Summand des Gesamtdruckes ist. Ebenso gültig ist die Betrachtung, dass jede Molekülsorte das Teilvolumen V i einnimmt, dass nun aber der Druck der Molekülsorte gleich dem Gesamtdruck p ist.
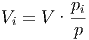 | (2.9) |
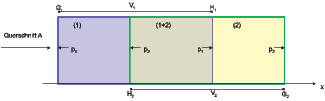
Reversible Entmischung mit teildurchlässigen Membranen
Wir betrachten als Gedankenexperiment das Teilsystem aus der Abbildung 2.24.2. Die beiden Volumina V 1 und V 2 sollen gegeneinander verschiebbar sein. Die Membrane (Wand) H1, die das Volumen V 1 nach rechts abschliesst, soll für die Molekülsorte 2 durchlässig, für die Sorte 1 aber undurchlässig sein. Ebenso soll die Membrane H2 für die Molekülsorte 1 durchlässig und für die Sorte 2 undurchlässig sein. Solche halbdurchlässigen Membranen existieren, zum Beispiel in Zellen. Hier wollen wir annehmen, dass die Membranen für die Molekülsorte, für die sie durchlässig ist, wie nicht-existent sein soll. Die beiden Membranen H1 und H2 bewirken also, dass sich im Überschneidungsgenbiet der beiden Volumina V 1 und V 2 beide Molekülsorten befinden.
Wenn man die beiden Volumina auseinander bewegt, so erzwingen die halbdurchlässigen Membranen, dass die Molekülsorten 1 und 2 getrennt werden. Wir wollen nun die beiden Zylinder von aussen um dx auseinander bewegen. Dabei soll der Zylinder 1 ruhend sein.
| Wand | Druck | Weg | Arbeit |
| H2 | p2 | -dx | dW1 = -Ap2dx |
| G2 | p2 | dx | dW2 = Ap2dx |
| H1 | p1 | 0 | dW3 = 0 |
| G1 | p1 | 0 | dW1 = 0 |
Die gesamte mechanische Arbeitsleistung ist dabei
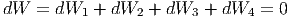 |
Die Entmischung benötigt also keine Energiezufuhr. Die innere Energie unseres abgeschlossenen, isolierten Gesamtsystems ändert sich nicht. Deshalb kann sich das System nie aus dem Gleichgewicht entfernen. Die Entmischung (und damit auch die Mischung) verlaufen reversibel. Für die Entropie gilt andererseits
| S | = S1 + S2 | ||
| S(T,V ) | = S1(T,V ) + S2(T,V ) | (2.10) | |
| S(T,p) | = S1(T,p) + S2(T,p) | (2.11) |
Bei m Komponenten jeweils mit der Molzahl νi ist die Gesamtentropie
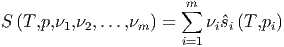 | (2.12) |
wobei ŝi die Entropie pro mol der Komponente i ist.
Wir betrachten m Komponenten mit den Molzahlen νi und der Temperatur T, die getrennt sein sollen. Der Druck jeder Komponente i wird durch isotherme Kompression auf den gewünschten Druck des Gesamtsystems, p, gebracht. Die jeweiligen Teilvolumina sind dann V i. Die Gesamtentropie S0 der m getrennten Teilsysteme ist einfach die Summe der Teilentropien.
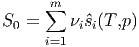 | (2.13) |
(alle Teilsysteme haben die gleiche Temperatur und den gleichen Druck!) Wir bringen nun die Teilsysteme zusammen und entfernen die Trennwände. Da der Druck und die Temperatur in jedem Teilsystem gleich sein sollen, bewirkt die Entfernung der Trennwände eine Durchmischung der Teilkomponenten, ohne dass aber bei diesen idealen Gasen die Temperatur oder der Druck sich ändert. Das neue Volumen ist V = ∑ V i. Die Teilkomponenten haben nun im Volumen V den Partialdruck pi. Die Entropieänderung durch die Diffusion ist
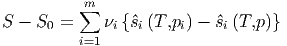 | (2.14) |
Aus unseren früheren Überlegungen mit dem Überströmen eines Gases aus einem Kompartiment in zwei wissen wir, dass
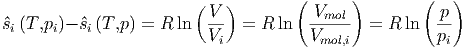 | (2.15) |
ist. Gleichung (2.14) kann mit den Partialdrucken pi als
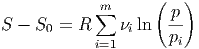 | (2.16) |
geschrieben werden. Aus Gleichung (2.16) folgt sofort, dass bei der Mischung die Entropie steigen muss, da aus p > pi folgt dass ln(p∕pi) > 0 ist und damit S - S0 > 0.
| Die Diffusion ist ein irreversibler Prozess! |
Die Verhältnisse des Gesamtdrucks zu den Partialdrucken sind proportional zu den Verhältnissen der Gesamtteilchenzahl zu den Teilchenzahlen der Teilkomponenten
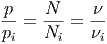 |
Damit ist die Entropiezunahme bei der diffusiven Mischung von m Teilchenzahlen
![( ) [ ]
∑m ν- m∑
S - S0 = R νiln ν = R ν ln ν - νilnνi
i=1 i i=1](td-20151236x.png) | (2.17) |
Wenn wir nun zwei Volumina voll mit gleichartigen Molekülen mischen, müsste natürlich
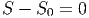 |
sein. Dazu müssen wir einen Grenzübergang betrachten,d er zwei unterschiedliche Molekülsorten kontinuierlich zu einer Sorte macht. Dieser Grenzübergang ist offensichtlich nicht machbar und unsinnig (Gibbsches Paradoxon).
Aus der Unmöglichkeit des kontinuierlichen Übergangs muss man schliessen, dass Atome nur in diskreten Zuständen vorkommen können.
Beispiel:
Edelgase als beste Approximationen des idealen Gases zeigen die Mischungsentropie. Sogar Orthowasserstoff und Parawasserstoff (zwei Spinzustände) sind nicht ohne Entropiezunahme mischbar.
Bei Systemen mit mehreren Molekülesorten sind die Anzahlen Ni thermodynamische Zustandsgrössen. Wir hatten früher die Entropie als Funktion der inneren Energie und des Volumens betrachtet:
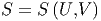 |
Nun müssen die Entropie als Funktion von innerer Energie, Volumen und m Teilchenzahlen betrachten.
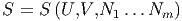 | (2.19) |
Wir nehmen zuerst an, dass alle Ni konstant sind. Dies ist gleichbedeutend, dass keine chemischen und kernphysikalischen Reaktionen vorkommen. Das Differential der Entropie war durch
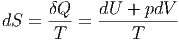 | (2.20) |
gegeben. Die partiellen Ableitungen der Entropie sind dann
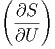 V ,N V ,N | = 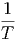 | ||
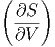 U,N U,N | = 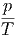 | (2.21) |
Unsere Diskussion der verallgemeinerten Kräfte legt nahe, dass zur extensiven Grösse Ni eine intensive Grösse μi gehören muss. Wir definieren
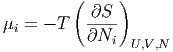 | (2.22) |
Hier bedeutet der Index N bei der Differentiation, dass alle Nj mit j ⇔ i konstant gehalten werden.
Das vollständige Differential der Entropie ist also
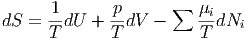 | (2.23) |
Je nachdem, welche Randbedingungen wir für das thermodynamische System verwenden, müssen wir das dazugehörige thermodynamische Potential verwenden. Daraus folgt
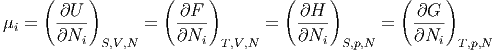 | (2.24) |
Alle thermodynamischen Potentiale sind extensive Grössen.
Zum Beispiel gilt für die freie Enthalpie G = G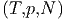 bei
nur einer Substanz
bei
nur einer Substanz
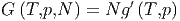 | (2.25) |
wobei g′ die freie Enthalpie pro Teilchen ist. Wir können aber auch schreiben
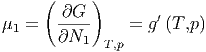 | (2.26) |
| Das chemische Potential μ ist die freie Enthalpie
pro Teilchen oder die freie Enthalpie pro
Molekül. |
Aus der Definition der inneren Energie
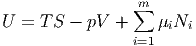 |
bekommen wir ganz allgemein das totale Differential
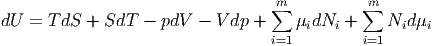 | (2.28) |
Andererseits wissen wir, dass U(S,V ,Ni) ist. Das Differential kann nur mit den freien Variablen gebildet werden.
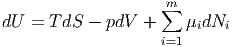 |
Deshalb muss die Summe der in Gleichung (2.28) für das totale Differential der inneren Energie nicht verwendeten Summanden identisch null sein. Dies führt auf die Gibbs-Duhem-Gleichung.
(Siehe Sommerfeld, Thermodynamik und Statistik [Som77, pp. 71-77])
Für den Fall, dass neben den intensiven Grössen Druck p und Temperatur T und den dazu gehörigen extensiven Grössen innere Energie U und Volumen V weitere extensive Grössen xi mit den dazu gehörigen verallgemeinerten Kräften Xi vorhanden sind, kann man den ersten und zweiten Hauptsatz zusammenfassen. Die Grössen xi sind allgemein. Die Teilchenzahl Ni ist nur ein Beispiel.
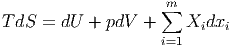 | (2.30) |
Bei variablen Teilchenzahlen ist xi = Ni und die dazugehörigen verallgemeinerten Kräfte Xi = -μi. Wir haben also
| TdS | = dU + pdV -∑ i=1mμ idNi | ||
| dU | = TdS - pdV + ∑ i=1mμ idNi | (2.31) |
Analog bekommen wir
| dH | = dU + d(pV ) | = TdS + V dp + ∑ i=1mμ idNi | (2.32) | |
| dF | = dU - d(TS) | = -SdT - pdV + ∑ i=1mμ idNi | (2.33) | |
| dG | = dU + d(pV ) - d(TS) | = -SdT + V dp + ∑ i=1mμ idNi | (2.34) |
Daraus bekommen wir durch Ableiten
| μi | = 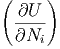 S,V ,Nj⇔i S,V ,Nj⇔i | (2.35) |
| μi | = 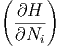 S,p,Nj⇔i S,p,Nj⇔i | (2.36) |
| μi | = 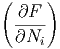 T,V ,Nj⇔i T,V ,Nj⇔i | (2.37) |
| μi | = 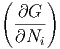 T,p,Nj⇔i T,p,Nj⇔i | (2.38) |
Analog zu früher können wir weiter Maxwellrelationen aus den zweiten Ableitungen bestimmen. Betrachten wir G.
| dG | = 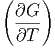 p,NjdT + p,NjdT + 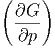 T,Njdp + ∑
i=1m T,Njdp + ∑
i=1m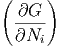 T,p,Nj⇔idNi T,p,Nj⇔idNi | ||
| = -SdT + V dp + ∑ i=1mμ idNi | (2.39) |
Daraus ergibt sich zum Beispiel
| S | = -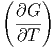 p,Nj p,Nj | (2.40) |
| μi | = 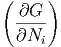 T,p,Nj⇔i T,p,Nj⇔i | (2.41) |
Die zweiten Ableitungen müssen von der Reihenfolge unabhängig sein, also
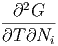 | = 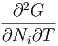 | ||
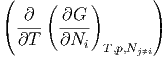 p,N
j p,N
j | = 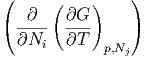 T,p,N
j⇔i T,p,N
j⇔i | ||
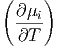 p,Nj p,Nj | = -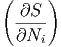 T,p,Nj⇔i T,p,Nj⇔i | (2.42) |
Wir betrachten nun ein Zweiphasensystem als ein System aus zwei Komponenten N1 und N2. Zum Beispiel könnte N1 die Anzahl Teilchen von CO2 in der Gasphase und N2 die Anzahl der CO2-Teilchen in der Flüssigphase sein.

Zweiphasensystem als System zweier Komponenten
Es gilt:
| U1 + U2 | = U = const. | ||
| V 1 + V 2 | = V = const. | ||
| N1 + N2 | = N = const. | (2.43) |
Weiterhin muss die Entropie
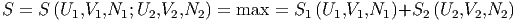 | (2.44) |
maximal sein
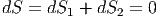 | (2.45) |
Aus Gleichung (2.43) folgt aber auch
| dU1 + dU2 | = 0 | dV 1 + dV 2 | = 0 | dN1 + dN2 | = 0 | (2.46) |
Wir führen die Differentiation aus und erhalten
| dS | = 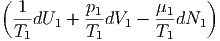 + + 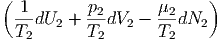 = 0 = 0 | ||
= 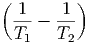 dU1 + dU1 + 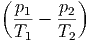 dV 1 - dV 1 -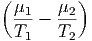 dN1 dN1 | (2.47) |
Wie immer müssen die Vorfaktoren der einzelnen Differentiale einzeln verschwinden. Daraus folgt
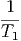 - -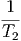 | = 0 | ⇒ | T1 | = T2 | |||||||
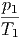 - -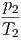 | = 0 | ⇒ | p1 | = p2 | |||||||
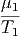 - -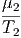 | = 0 | ⇒ | μ1 | = μ2 | (2.48) |
Wir kommen also zur Aussage:
| Im Phasengleichgewicht sind Temperatur,
Druck und die chemischen Potentiale gleich. |
Bei reinen Phasen gilt auch: g1 = g2 (Gleichheit der freien Enthalpie pro Molekül)
Sind verschiedene Teile eines Gesamtsystems im Gleichgewicht, gilt allgemein
μ1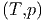 | = μ2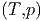 | (2.49) |
 Lizenzinformationen
Lizenzinformationen
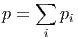
![[ ∑m ]
S - S0 = R ν ln ν - νiln νi
i=1](td-20151237x.png)
![[μ]](td-20151247x.png)